PROTON EXCHANGE MEMBRANE FUEL CELLS AS AN ELECTRICAL POWER SOURCE: RECENT DEVELOPMENTS AT THE UNIVERSITY OF KANSAS
by
Trung Van Nguyen
Department of Chemical and Petroleum Engineering
The University of Kansas
Lawrence, Kansas 66045
913-864-3938 (Phone)
913-864-4967(Fax)
[email protected]
Abstract
Proton Exchange Membrane (PEM) fuel cells are becoming more popular as an energy conversion device because its efficiency and simplicity in design and operation. Significant improvements and recent developments in the areas of water and heat management, membranes, electrode designs, and, most recently, the interdigitated flow field design, have increased greatly the attractiveness of this system. However, further improvements in the areas of performance, efficiency and costs are needed to make this system more competitive. High-power-density PEM fuel cells are being developed at the University of Kansas using new electrocatalyst supported materials, thinner and more conductive metal bipolar plates, an approach in which gas plasma etching is used to enhance the interfacial area between the membrane and the electrode, the recently developed interdigitated flow field design, and direct liquid water injection for membrane humidification. Results obtained show significant performance improvements and processing costs reduction.
Introduction
With increasing environmental concerns over vehicle generated pollution and the limited range associated with battery-powered electric vehicles, the Proton Exchange Membrane (PEM) fuel cell system is gaining more attention as an alternative power generation source for electric vehicles. Its attractive characteristics include high efficiency, simple design and operation, self starting at low temperatures, low cost construction materials and comparable driving range and refueling time to conventionally powered vehicles. However, before the PEM fuel cell system can become economically competitive with existing technologies and be applied in wide-range practical situations, its power density must be increased by a factor of six and its costs reduced by a factor of 10 [1].
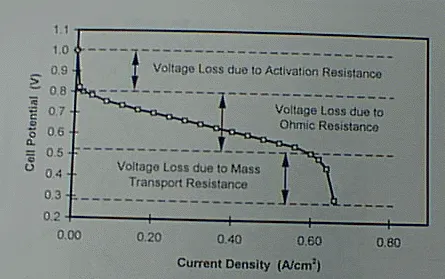
Areas for improvement in the performance can be identified by evaluating the polarization (E vs. I) curve of a fuel cell (See Figure 1). This curve can be separated into three regions: 1) the very sharp voltage drop in the first region associated with the activation resistances of the cell, 2) the gradual drop in the voltage in the second region attributed to the ohmic resistances in the cell, and 3) the second sharp voltage drop in the third region attributed to the mass transport resistance in the cell.
The sharp voltage drop of the first region associated with the activation resistance is attributed to the type of catalyst and the active catalyst surface area that is in contact with the electrolyte and the electrical network in the electrode and is accessible to the reacting gases. Lowering this resistance will raise the whole polarization curve. This voltage loss can be reduced by using catalysts with lower activation resistance and increasing the catalyst surface available for reaction per unit volume of electrode. Currently, platinum is the best catalyst available. Prior efforts in sputtering the catalyst onto the surface of the electrode that is later hot pressed onto the membrane and mixing the catalyst-substrate with the membrane ionomer have resulted in significant improvement in the fuel cell performance and reduction in catalyst loading. Our electrocatalyst development approaches at KU will result in further reduction in the catalyst loading, lower materials processing costs and better electrode and membrane stability.
The gradual drop in voltage of the second region known as the ohmic voltage loss is attributed to the ohmic resistance of the components within the electrical network of the fuel cell such as the electrical and contact resistances of the electrodes, membrane and current collecting components. Lowering this resistance will result in a lower slope in the E-I curve and consequently higher power densities at higher energy efficiencies. Voltage loss in the second region has been successfully reduced by humidifying the reactant gases, especially the anode gas, as a result of better understanding of the coupling effect of proton transport and water transport in the membrane; and employing thinner membranes and membranes with lower ionic and water transport resistances. Component contact resistances, however, have not been addressed. Our use of thinner and more conductive metal components and the liquid water injection strategy are designed to reduce the voltage loss in this region.
Finally, the sharp voltage drop of the third region known as the voltage loss associated with the mass transport resistance is attributed to concentration polarization, which occurs as a result of the depletion of the reactant at the reaction interface as its transport to the reaction sites fails to keep up with the reaction rate. This phenomenon is especially severe at the cathode of the fuel cell where oxygen is the reactant. The concentration of oxygen at the reacting surface is not only depleted by the reaction by also by the creation of an additional barrier to its transport to the surface by the presence of liquid water, the product of the cathodic reaction and proton transport from the anode, on the catatyst/membrane surface and within the porous structure of the electrode. Minimizing this resistance will allow the ohmic region to be extended and result in much higher power density operation. The voltage loss in this region has not been fully addressed. So far it has been solved indirectly by raising the gas pressure (increasing the local concentrations) and partially by raising the gas stoichiometric flow rates which results in lower energy efficiencies. The interdigitated flow field recently developed by this author [2] is design to address this problem directly.
Efforts to Reduce Activation Resistances
A PEM fuel cell is a solid-electrolyte based system. Consequently, old concepts associated with a liquid-electrolyte based system are no longer applicable. Since its electrolyte is a solid and not a liquid, it does not penetrate into the micropores of the electrode particles even in the case when the electrode is made of a catalyst supported substrate that is pre-mixed with the membrane ionomer. Consequently, the traditional electrode preparation technique of dispersing very fine platinum particle (50A° ) within the carbon particles does not yield any significant improvement in the catalyst active surface area. It is this authors strong belief that only the catalyst area on the outer surface and on the wall of the macropores of the substrate in contact with the ionically conductive membrane and accessible to the reactant gas that is truly active. The rest is wasted. There have also been works in which platinum was deposited on or near the membrane surface [3]. In this case, only the catalyst on the surface is useful. Catalyst within the membrane that is not in electrical contact with the current collector is again also wasted. My research group at the University of Kansas is currently evaluating the deposition of platinum on a fine carbon powder substrate as an alternative catalyst-supported material for electrodes for PEM fuel cells. Figures 2 and 3 show scanning electron micrographs (under the same magnification) of the standard commercially available platinum black catalyst and our catalyst. Note the difference in the morphology and higher surface area of our finer catalyst. This catalyst is undergoing testing first by cyclic voltammetry and then in actual fuel cells.
In addition to the development of a more cost effective catalyst for electrodes for PEM fuel cells, the author is also exploring the use of the gas plasma etching technique used in processing electronic materials as a way to create a 3D pattern that will increase the external surface area of the membrane. This process is expected to have great promises because of its well defined and very fine pattern nature and the fact that it is a well practiced process in the electronic materials processing industry. If this approach is successful, two possibilities will result. First, by creating a 3D surface on the membrane deeper penetration and higher contact area between the membrane and the catalyst substrate will be achieved during hot pressing of the membrane onto the electrode. Second, if sufficient surface area can be created with the etched patterns, platinum catalyst can then be deposited directly onto the membrane thereby eliminating the hot pressing of the electrode onto the membrane step and the need for a binder to hold the catalyst particles together. Furthermore, the stronger adhesive strength resulting from the deposition process as compared to that achieved by the hot pressing technique will reduce if not eliminate completely the contact resistance at the electrocatalyst/membrane interface and future delamination problem frequently encountered as a result of the hydration/dehydration cycles during prolong operation. This work is currently underway.
Efforts to Reduce Membrane and Mass Transport Resistances
Recent experimental analysis has shown that a delicate balance exists between the reactive and transported species and the hydration state of the polymer electrolyte within the membrane/electrode assembly (MEA). At moderate to high operating current densities, the rate of back-diffusion of water from the cathode is often exceeded by the rate of electro-osmosis from the anode. This situation can result in membrane dehydration, thus making it highly resistive, in a very short period of time. An equally important problem is that of cathode flooding from proton transport and water generation. Therefore, it is necessary to keep the membrane properly hydrated to ensure that it maintains high conductivity while simultaneously controlling water accumulation at the cathode.
There are several methods of humidifying a PEM fuel cell [4]. The method my research group at KU has been focusing on is liquid water injection directly into both the anode and cathode reactant gas streams. We have also done a comparison test to validate further the effectiveness of the interdigitated flow distribution design that I have developed over that of the conventional one [2].
The results from these tests are summarized in Figures 4, 5 and 6. They show that the advanced interdigitated flow field has yielded performance improvements of up to 100% and higher over the conventional design. The interdigitated design utilizes dead-ended channeling which changes the predominant transport mechanism from diffusion to convection and is the main reason for the dramatic increase in performance.
Large increases in performance have also been obtained with liquid water injection, particularly into the anode gas stream. It has been found that the cathode layer is more sensitive to liquid water injection and is more easily flooded than the anode layer. Figures 4 and 5 show that because of its higher volumetric flow rate the anode can handle much higher liquid water flow rates than the cathode before flooding at the respective electrode occurs. This trend has been observed using both types of flow fields.
Another important finding is that liquid water injection used with the interdigitated flow fields gives a larger performance increase than does liquid water injection used with the conventional flow fields. This phenomenon is probably because the dead-ended channeling causes a constant water supply for proton migration to be maintained within the anode layer and aids in excess water removal from the cathode layer. Similar data have been obtained using either air or oxygen as the cathode gas.
A strong relationship between the mass flow rates of reactant gases and liquid water flow rates has been observed as well. Experimental data has shown that lower operating pressure, higher mass flow rate of reactants, and air as the cathode gas all enable higher liquid water flow rates to be used before flooding occurs or the maximum performance level is achieved (Figure 6 vs. Figure 4). These findings indicate that the hydrodynamics of the reactant gases are closely tied to the amount of liquid water a PEM fuel cell can handle. Figure 6 shows some performance curves obtained using air as the cathode reactant.
Acknowledgments
The author would like to thank the University of Kansas, the State of Kansas, and the K*STAR NSF-EPSCoR program for the financial support of this work.
References
Figure 1. Typical polarization curve for a PEM fuel cell.

Figure 2. Scanning electron micrograph of 20% Pt commercial catalyst (on Vulcan XC-72)\

Figure 3. Scanning electron micrograph of KUs 20% Pt deposited on carbon black
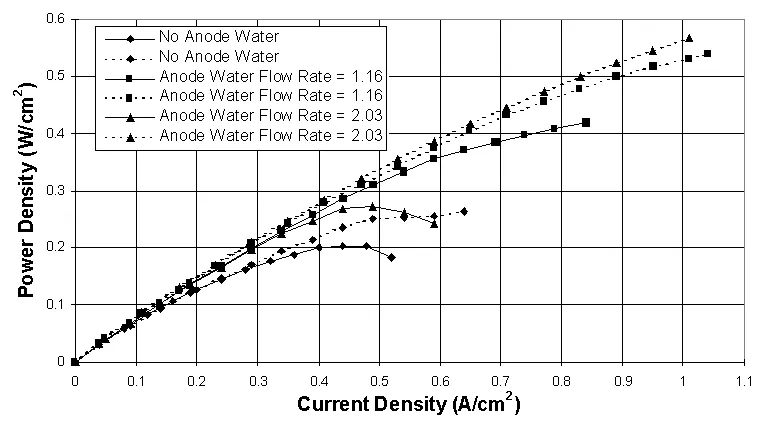 Figure 4. Comparison between conventional (solid lines) and interdigitated (dashed lines) flow fields using H2/O2 at 80° C, 1 atm abs., and several anode water injection levels. Anode water flow rates are given in mol H2O/mol H2, and the cathode water flow rate is 0.635 mol H2O/mol O2. Gas flow rates used are 2.50 A/cm2 equiv. H2 and 3.00 A/cm2 equiv. O2.
Figure 4. Comparison between conventional (solid lines) and interdigitated (dashed lines) flow fields using H2/O2 at 80° C, 1 atm abs., and several anode water injection levels. Anode water flow rates are given in mol H2O/mol H2, and the cathode water flow rate is 0.635 mol H2O/mol O2. Gas flow rates used are 2.50 A/cm2 equiv. H2 and 3.00 A/cm2 equiv. O2.
 Figure 5. Comparison between conventional (solid lines) and interdigitated (dashed lines) flow fields using H2/O2 at 80° C, 1 atm abs., and several cathode water injection levels. Cathode water flow rates are given in mol H2O/mol O2, and the anode water flow rate is 0.361 mol H2O/mol H2.
Figure 5. Comparison between conventional (solid lines) and interdigitated (dashed lines) flow fields using H2/O2 at 80° C, 1 atm abs., and several cathode water injection levels. Cathode water flow rates are given in mol H2O/mol O2, and the anode water flow rate is 0.361 mol H2O/mol H2.
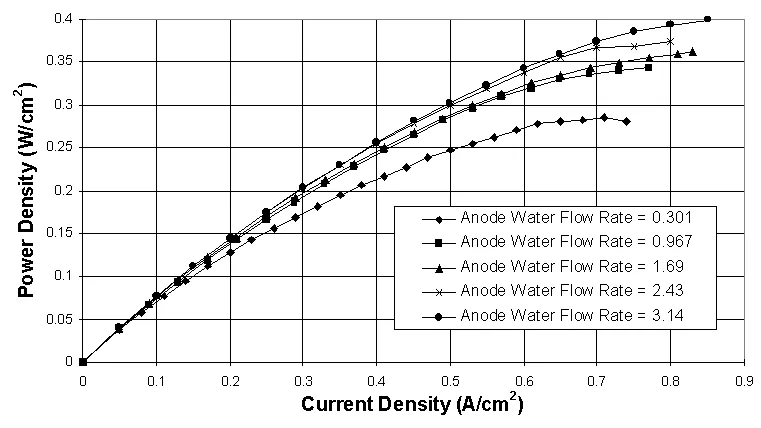
Figure 6. Anode liquid water injection performance study using interdigitated flow fields and H2/Air at 60° C, 1 atm abs., and various anode water injection levels. Anode water flow rates are given in mol H2O/mol H2, and the cathode water flow rate is held constant at 0.200 mol H2O/mol Air. The gas flow rates are 3.00 A/cm2 equivalent H2 and 2.00 A/cm2 equivalent O2.